So let’s have some good antibiotic news to start the week – that’s always welcome, given that bacteria don’t take weekends or holidays themselves. This new paper describes a synthetic variant on clindamycin, which is itself a synthetic variant (chlorination!) of the natural product lincomycin. Clindamycin (on the left in the illustration below) is pretty strong stuff already; it’s given mostly for infections caused by anaerobic bacteria (Gram-positive and Gram-negative), where it works by inhibiting protein synthesis. Bacteria aren’t the only organism it kills – when combined with cholorquine, it’s also an effective malaria treatment. But if you’re taking it, it should be for a good reason, becuase it increases the chances of intestinal Clostridium difficile infections even compared to treatment with other broad-spectrum antibiotics, where that can also be a problem. “C diff”, an anaerobe that’s not as susceptible to clindamycin, can overrun the colon tissue after the gut microbiota are severely disturbed, and the infection is both very unpleasant (at the very least) and difficult to shake. There are other organisms that clindamycin has trouble with as well, such as Pseudomonas and Enterobacter species.
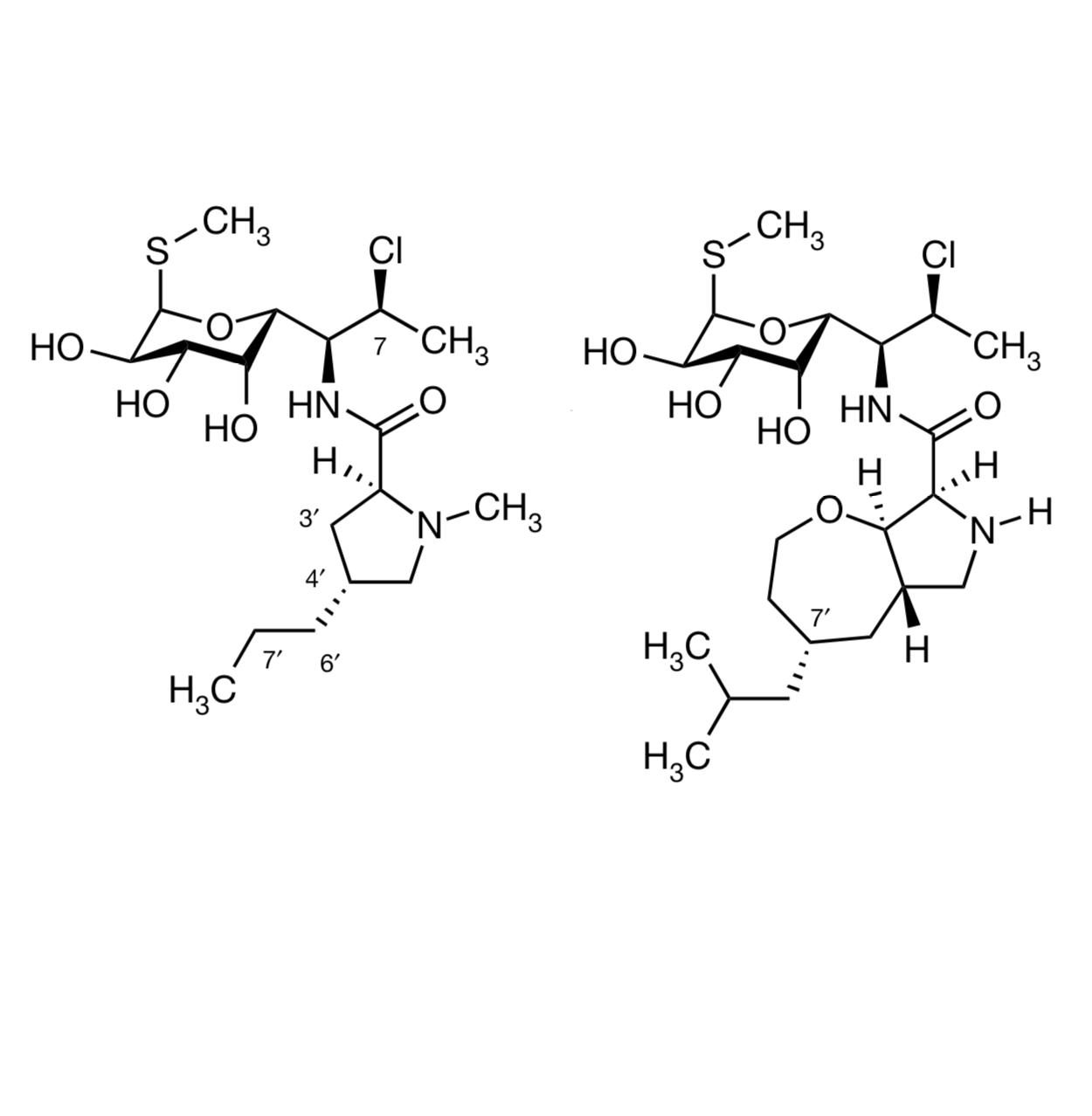
This latest work is a collaboration between the Myers group at Harvard and the Polikanov group at UI-Chicago, and it involves making about 500 variations on that substituted proline. As you’d guess from the protein synthesis mode of action, clindamycin binds to the bacterial ribosome, and there are X-ray structures that show that propyl chain as important for binding. So most of the analogs in this work focused on variations in that region that modeled well, with lots of bicyclic derivatives. The syntheses of these came out of earlier work from the Myers group on the synthesis of macrolide antibiotics, repurposed here to excellent effect.
Many of these showed similar or even somewhat better activity compared to the parent antibiotic, showing that the hypothesis was on the right track. It is quite easy to wipe out antibiotic efficacy if you pick the wrong idea and the wrong part of the molecule to modify, an effect which I (and many others who have worked in the field over the decades!) have verified many times by personal experience. The molecule at the right in the illustration, which has been named iboxamycin, was the most active of the bunch, though, and the group went on to work out a new synthesis of it that could provide multigram amounts.
The compound shows a really impressive range of activity, notably wider than clindamycin and effective against a horrible lineup of resistant bacteria. This included various Streptococcus, Staphylococcus, Enterococcus, Klebsiella, Acenitobacter and others, with a lineup of strains that show resistance to treatment (in various species and strains) to the parent clindamycin and to methicillin, vancomycin, linezolid, doxycycline, azithromycin, levofloxacin, tetracyclines, aminoglycosides, cephalosporins, etc. It is also much more potent against Clostridium difficile, which suggests that it might be less prone to letting that one take over the colon (although that certainly has to be confirmed in the clinic eventually. It was not toxic to human cells, and in mice, it has better PK and blood levels than clindamycin itself. It was active in models of both localized infection (with several resistant bacterial strains) and in systemic infection in a lethal challenge with S. pyogenes.
The team obtained an X-ray structure of the compound bound to the 70S ribosome subunit, and it does indeed sit in the same pocket as clindamycin. Functionally, it seems to bind with greater affinity, and that isobutyl group sits in a cleft of the ribosome that’s normally used to accommodate the side chains of incoming amino acids in protein synthesis. That’s where clindamycin’s propyl group is, but iboxamycin extends further into the P site, which might explain its greater ability to interfere with ribosome function. Another X-ray structure with ribosome subunits from a clindamycin-resistant strain suggested that these interactions persist, and are enough to overcome the mutation in these species that disrupts binding of the aminosugar part of the structure. In fact, the ability of the various derivatives to overcome this resistance mutation seems to correlate well with the length and size of the alkyl groups in that part of the molecule, which suggests a general route to better ribosomal targeting.
But lest you think that we’re ready to model our way to happiness, the authors specifically note that “our findings attest to the continued role of empiricism in antibiotics discovery, as the activity of oxepanoprolinamides against posttranscriptionally methylated ribosomes could not have been predicted on the basis of existing antibiotic–ribosome co-crystal structures“. It’s much easier to rationalize the results once you come across a great compound than it is to predict what that great compound will be! But this work suggests a number of possibilities for even further improvements, and who knows what will be discovered when those are tried out? Overall, these results are very encouraging indeed, both for this specific compound and for the whole project of coming up with new resistance-evading antibiotics. I’ve been really happy to see the powers of total synthesis being aimed in this direction. Back when I was doing total synthesis of macrolide antibiotics in the 1980s in grad school we paid lip service to this sort of thing, but in practice it generally didn’t happen (and it especially didn’t happen to the point of thinking about scale-up of the synthetic routes!) Somewhere, R. B. Woodward is raising a glass of Scotch and giving this field a round of applause.
