I’m talking about the improvement in cryo-electron microscopy to get down to atomic resolution. I’ve spoken about cryo-EM several times before on the blog (most recently here and also elsewhere), since over the lifetime of the blog itself the technique has made huge advances. Now two papers in Nature report the latest, which takes the quality of cryo-EM data to a level that twenty years ago not many people would have ever expected to see.
The early 2010s brought a “resolution revolution” that isn’t over yet. It’s been a combination of several techniques and improvements in both hardware and software. Cryo-EM can be computationally intensive, so even if the hardware had been available in (say) 1985, we wouldn’t have been able to extract the information out of it that we can now (at least not in any useful amount of time!) But it’s not all just better processors and better software; the newer hardware is no small part of the story. We have better electron sources, better ways to keep the energies of the beam within a very narrow range and very precisely aimed, better detectors at the back end of the sample, new noise-reduction techniques in general – all of these and more have combined with all that software power to produce something amazing.
If you’re wondering what the big deal is, it might be summed up as “you don’t always have to grow crystals any more”. X-ray crystallography has been a huge technique ever since its beginnings early in the 20th century, and has advanced over the decades for some of the same reasons that cryo-EM has more recently: better X-ray sources (both brighter and tighter), better detectors, and better algorithms and hardware to run them on. Modern crystallography would floor the folks who were doing huge amounts of work to generate every single structure back in the 1950s. But you’ve still needed crystals, high-quality ones, to get the technique to work. And growing them is witchcraft. Really, it is. As I’ve said many times, all you have to do is walk into a protein crystallography lab and see the stacks of multiwell plates everywhere, full of dozens, hundreds, thousands of attempts to grow good crystals by varying the concentrations, varying the buffers, varying the protein constructs, varying the long, long list of salts and counterions and additives that people have tried over the years to coax the proteins into somehow lining up and forming orderly ranks.
As anyone who’s done the slightest bit of work in the field will attest, even once you see beautiful chunky crystals forming – and not those fluffy threads, those usually don’t work out – you’re nowhere near out of the woods. The first big question is “Is that actually your protein?” because those salts and additives can form nice crystals of their own, too, and the second big question is “Does it diffract?” Very decorative-looking crystals can turn out to be crap when the X-ray beam hits them: what you want is showers of well-defined diffraction spots, varying in sweeps of beautiful patterns as the orientation changes between the crystal and the beam, and stretching way out to the edges of your detector (the further out they go, the higher the resolution you will likely be able to wring out of the eventual data set). What you get, too often, is splorts and splotches, smeary gorp that starts out ugly and gets uglier as you watch. Robert Palmer warned us years ago that a pretty face don’t mean no pretty heart, and every crystallographer knows that he was right. Why yes, I have tried growing crystals and collecting data on them personally: did I give myself away?
But what if you didn’t have to do this? Cryo-EM doesn’t need crystals. You take your protein of interest and spread a solution of it out on a surface, which after a few more steps gives you a scattering of individual protein particles lying there every which way. And you hit them individually with your fancy electron beam and collect the results on your fancy detector, with all that data going into some very fancy software indeed. That software can, if all goes well, sort the particles you collected on into categories – edgewise, tilted, right down the middle, lying on the long side, and so on – and build a model of what the protein must look like that could have generated the data. Used to be, this process gave you fuzzballs, but they’ve been getting relentlessly sharper.
And now we’re down to individual atoms, which is what a high-quality X-ray structure has always been able to deliver. As the resolution of these techniques improves, the pictures become startling. An aromatic ring, for example, like a phenylalanine side chain on a protein, is a fuzzy blob with lousy data. Better data show you that it’s a flattened blob, and you can start to see how it’s tilted in three dimensions. Produce higher-quality stuff, and that aromatic ring stops looking like a hexagonal lump of wood and starts becoming a hexagonal doughnut: you’re seeing the open space in the center of the six-membered ring. That level is very nice resolution for a protein structure, but the small-molecule crystallographers can push on to even more. Higher resolution still, and that doughnut turns into a ring made from six ball bearings: the individual carbon atoms. Beyond that, you can even start to see ghostly electron density between them, which is the shared electrons of the chemical bonds themselves. You can see the improvement in the real-world structures below, from this review article. The ball-and-stick stuff is the model, and the meshwork is what you actually get:
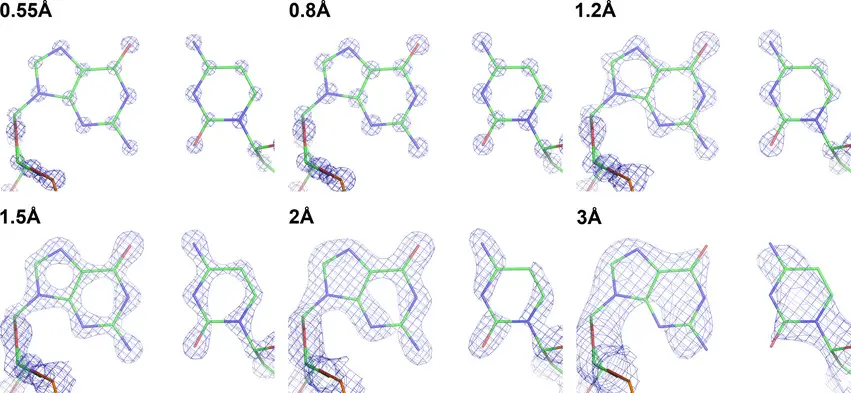
Note that the highest-resolution structures there are rare and difficult. But these two new cryo-EM papers have a well-behaved protein (apo-ferritin) resolved to just over 1.2 Ångstrom, and a membrane protein (a GABA receptor) at 1.7Å. That’s really damned good, and the exciting thing is that the technology is still improving. This overview at Nature suggests that we may be getting near the end improving the electron beams and the like, but that there’s plenty of room in sample preparation and data analysis. This will allow us to see more and more detail on progressively harder and harder to handle samples.
Consider cryo-electron tomography, where you apply these techniques to proteins in the cells themselves. This is a pretty intense technique, but it’s already providing insights into protein structure and function that we couldn’t get any other way. This stuff would have been considered impossible not all that long ago, and I’m very happy to be able to see it happening for real.
