I feel like a dose of good ol’ organic chemistry this morning, and a (virtual) meeting I attended yesterday gave me a paper to talk about that delivers some. I was speaking with a local group of modelers and computational chemists (BAGIM), and MIT’s Connor Coley was there presenting some of his group’s work.
I had missed this preprint from him and Wenhao Gao, which I’d like to highlight today. It’s on the broad subject of using computational methods to come up with plausible small molecules against a given biological target, which is one of those things that everyone would like to do and no one can quite be sure that they’re able to manage yet. There are plenty of ways to approach the problem, and these range from “indistinguishable from trivial” all the way to “indistinguishable from magic”.
As you’d guess, the magic-powers end of the scale tends to be both computationally intensive and wildly variable in its results, and this is the place to say what I often say about such things. That is, I’m a short-term pessimist and a long-term optimist. I see no reasons why we shouldn’t be able – eventually – to compute our way to fresh drug structures. But at the same time, I think that we have to get a lot better at it than we are now. This is a really hard problem, as has been abundantly demonstrated for several decades now, and it’s hard for a lot of hard reasons. Let’s list a few!
First, the compounds themselves can range over a potential chemical space that is far too large for the human mind to comprehend, and far too large to calculate through in any direct fashion. I mean that in the most literal sense – our solar system has not been around long enough to furnish us the the time to evaluate all those possible structures. And that’s just the structures as received. In real life, they can often adopt all sorts of three-dimensional conformations, and their interactions with protein targets can make them choose some that you wouldn’t have expected. That’s because those interactions are many and various themselves, including but not limited to hydrogen bonds, interactions between pi-electron clouds, van der Waals interactions, halogen bonds, and many more. Our abilities to model these computationally is. . .evolving, let’s say. And the proteins that such compounds bind to have a range of possible pockets and surfaces that are just as wide-ranging as the small-molecule structures are, and that includes their conformational flexibility as well. Add in the effects of key water molecules around these sites, which can be contacted or displaced to completely change the thermodynamics of ligand binding, and you have plenty to deal with.
Speaking of those thermodynamics, surprisingly small energy differences will determine if a given molecule binds to its target or not – differences that are very likely within the error bars of many of your calculations. Those calculations, in turn, will tell you little or nothing about the on-rates and off-rates of that binding process, and that can be yet another huge factor in whether or not you have an interesting compound or not. Dealing with such molecular dynamics is another kettle of hostile, uncooperative fish and can call on computing resources that will strain the largest systems you can possibly access without needing to make contact with intelligent space aliens.
But here’s where I flip around and say again that none of these look like insoluble problems. There seem to be no mathematical or chemical reasons why we can’t get a better handle on them, given sufficient time, money, effort, and ingenuity. It breaks no laws of physics to be able to calculate the binding of a compound to a protein target well enough to determine its affinity as accurately as an assay in a few microliters of solvent can, and to do that over and over with a varied list of compound structures isn’t impossible, either. Just really hard.
One hard limit you do bang into though, as mentioned, is the sheer size of chemical space. We need algorithmic help in navigating that, and that’s the subject of the preprint linked above. It’s talking about “generative” methods to navigate through such space, an idea I last spoke about here. The concept is pretty easy to describe but pretty tricky to implement: you allow the software to build out molecules from some starting point, accepting and rejecting particular lines of inquiry so as to focus the computational resources on the richer seams of structural space. That is, you’re generating new structures in a deliberate fashion instead of just (impossibly) running through every variation possible.
These models need some breakfast in order to get out of bed, computationally speaking. One way is to train them on a set of molecules in order to generate some hypotheses about the chemical spaces in which they reside. If you don’t have any of those or don’t want to introduce possible biases that way, you can try letting some software loose to try to score newly generated molecules against a “black-box” function, but you are then running a larger risk of going on a computational snipe hunt. All sorts of approaches to both of these methods have been proposed.
But what sorts of molecules then get generated? In 2016, I took a look at a compound-generating effort that led to some pretty funky structures, and what I take away from the current preprint is that the funky beat continues. The new paper evaluates three generative algorithms (SMILES LSTM, SMILES GA, and Graph GA) with fourteen multi-property objective functions (which is how you try to convert a chemical structure into a numerical fitness score). These are the techniques used in the “Guacamol” paper on de novo compound design, proposed there as a benchmark for such efforts. Gao and Coley show, for example, that if you use the SMILES GA algorithm with the MPO that’s derived from the structure of the known EGFR drug osimertinib, the structure shown below is a top-scoring suggestion.
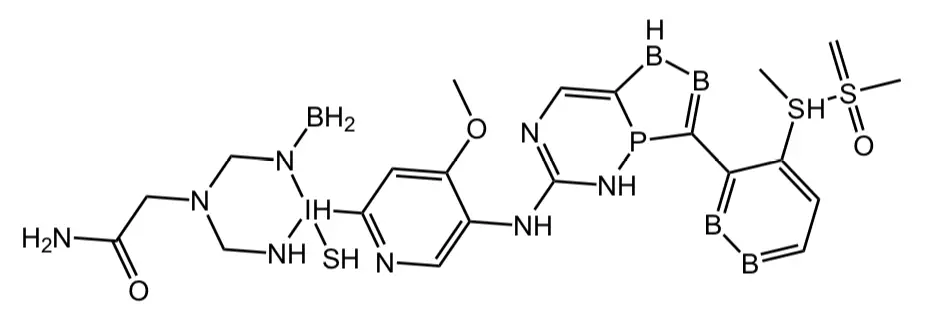
That’s clearly not a very useful suggestion. Admittedly, it’s the worst example in the paper, but that computational combination did not generate an actual make-able compound in any of its top 100 molecules. Looking through the other examples, all sorts of odd P, N, O and S functionality appear, ranging from inadvisable down to nutball – endoperoxides, hypochlorites, aromatic phosphorus rings, weirdo phosphines, four-membered rings with three nitrogen atoms in them, and so on.
So this preprint is attempting to impose some order on the situation with various suggestions for synthesizability calculations, with particular attention to MIT’s previously reported ASKCOS system in a paper I blogged about here. There are other ways of doing this, which are compared in the paper – for example, you could send the structures to a retrosynthesis program and see what it makes of them, but that’s a time and computing-cycle intensive way to go. Anyway, the take-home, at least for me, is that sometimes you get a pretty reasonable list of compounds and sometimes you don’t. The systems that train up on a set of known compounds tend to give you more realistic lists, whereas the “goal-oriented” ones that use a black-box function and bootstrap their way along tend to give you the wilder ones.
When you get one of those crazy lists, applying the synthesizability screen throws out so many compounds that the ones that are left may not even be anything that the algorithms found very interesting in the first place. On the other hand, sometimes you get lists right off the bat whose synthesizability scores are actually better than the one you get (68%) by running the ChEMBL database past the ASKCOS software – and remember, ChEMBL is made up exclusively of compounds that are actually known to have been made and tested. You could also apply some synthesizability rules up front, so weirdo molecules aren’t even generated in the first place, but how heavy a hand to use there is another question.
So we don’t have a perfect answer yet, naturally. But this preprint is a good start on the problem, and shows how large that problem is under some conditions. And it’s funny to see this play out, because long-time drug discoverers will have deja vu feelings about having had just these sorts of interactions with their modeling colleagues over the years. “The model says that you should do X”. “Well, we can’t do X – what else does the model say?” “Nothing that looks anything as good as if you could do X” And so on. Good to know that this well-established iteration has now been automated!
